Segway 1.1 documentation¶
| Homepage: | http://noble.gs.washington.edu/proj/segway |
|---|---|
| Author: | Michael M. Hoffman <mmh1 at uw dot edu> |
| Organization: | University of Washington |
| Address: | Department of Genome Sciences, PO Box 355065 Seattle, WA 98195-5065, United States of America |
| Copyright: | 2009-2011 Michael M. Hoffman |
| Last updated: | May 05, 2011 |
For a conceptual overview see the paper:
Michael M. Hoffman, Orion J. Buske, Zhiping Weng, Jeff A. Bilmes, William Stafford Noble. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Submitted.
Michael <mmh1 at uw dot edu> can send you a copy of the latest manuscript.
Quick start¶
Installation and configuration¶
To install Segway in your own user directories, execute this command from bash:
python <(wget -O - http://noble.gs.washington.edu/proj/segway/install.py)
The installer will ask you some questions where to install things and will install (if necessary) HDF5, NumPy, any other prerequisites, and Segway. It will also tell you about changes it wants to make to your ~/.bashrc to set up your environment properly.
Log out and back in to source the new ~/.bashrc.
If you are using SGE, your system administrator must set up a mem_requested resource for Segway to work. This can be done by installing Segway and then running python -m segway.cluster.sge_setup.
Acquiring data¶
Observation data is stored with the genomedata system. <http://noble.gs.washington.edu/proj/genomedata/>. There is a small Genomedata archive for testing that comes with Segway, that is used in the below steps. You can get it using:
wget http://noble.gs.washington.edu/proj/segway/2011/test.genomedata
Running Segway¶
Use the segway train command to discover patterns in the test data. Here, we specify that we want Segway to discover four unique patterns:
segway --num-labels=4 train test.genomedata traindir
Use the segway identify command to create the segmentation, which partitions the genome into regions labeled with one of the four discovered patterns:
segway identify test.genomedata traindir identifydir
Results¶
- The identifydir/segway.bed.gz file has each segment as a separate line in the BED file, and can be used for further processing.
- The identifydir/segway.layered.bed.gz file is designed for easier visualization on a genome browser. It has thick lines where a segment is present and thin lines where it is not. This is not as easy for a computer to parse, but it is more useful visually.
- You can also perform further analysis of the segmentation and trained parameters using Segtools <http://noble.gs.washington.edu/proj/segtools/>.
Installation¶
Segway requires the following prerequisites:
You need either Sun Grid Engine (SGE; now called Oracle Grid Engine), or Platform Load Sharing Facility (LSF) and FedStage DRMAA for LSF. You need Graphical Models Toolkit (GMTK), which you can get at <http://noble.gs.washington.edu/proj/segway/gmtk/gmtk-20091016.tar.gz>. You probably need to install NumPy separately.
You will need these Python packages, which will probably be installed automatically by pip install segway or easy_install segway: genomedata>0.1.5, textinput, optbuild>0.1.6, optplus>0.1.0, tables>2.0.4, forked-path, colorbrewer, segway, drmaa>=0.4a3.
If you are installing as an individual user, we have developed an install.py script that should make this easy.
If you are installing as a system administrator, we recommend using pip install segway or easy_install segway without configuring ~/.pydistutils.cfg to install in your home directory. This should install many of the prerequisites as well.
Cluster configuration¶
If FedStage DRMAA for LSF is installed, Segway should be ready to go on LSF out of the box.
If you are using SGE, someone with cluster manager privileges on your cluster must have Segway installed within their PYTHONPATH or systemwide and then run python -m segway.cluster.sge_setup. This sets up a consumable mem_requested attribute for every host on your cluster for more efficient memory use.
The workflow¶
Segway accomplishes four major tasks from a single command-line. It–
- generates an unsupervised segmentation model and initial parameters appropriate for this data;
- trains parameters of the model starting with the initial parameters; and
- identifies segments in this data with the model.
4. calculates posterior probability for each possible segment label at each position.
Technical description¶
More specifically, Segway performs the following steps:
- Acquires data in genomedata format
- Generates an appropriate model for unsupervised segmentation (segway.str, segway.inc) for use by GMTK
- Generates appropriate initial parameters (input.master or input.*.master) for use by GMTK
- Writes the data in a format usable by GMTK
- Call GMTK to perform expectation maximization (EM) training, resulting in a parameter file (params.params)
- Call GMTK to perform Viterbi decoding of the observations using the generated model and discovered parameters
- Convert the GMTK Viterbi results into BED format (segway.bed.gz) for use in a genome browser, or by Segtools <http://noble.gs.washington.edu/proj/segtools/>, or other tools
- Call GMTK to perform posterior decoding of the observations using the generated model and discovered parameters
- Convert the GMTK posterior results into bedGraph format (posterior.seg*.bedGraph.gz) for use in a genome browser or other tools
- Use a distributed computing system to parallelize all of the GMTK tasks listed above, and track and predict their resource consumption to maximize efficiency
- Generate reports on the established likelihood at each round of training (likelihood.*.tab)
The identify and posterior tasks can run simultaneously, as they depend only on the results of train, and not each other.
Data selection¶
Segway accepts data only in the Genomedata format. The Genomedata package includes utilities to convert from BED, wiggle, and bedGraph formats. By default, Segway uses all the continuous data tracks in a Genomedata archive.
Tracks¶
You may specify a subset of tracks using the --track option which may be repeated. For example:
segway --track dnasei --track h3k36me3
will include the two tracks dnasei and h3k36me3 and no others.
It is very important that you always specify the same --track options at all stages in the Segway workflow. There is also a special track name, dinucleotide. When you specify --track=dinucleotide, Segway will create a track containing the dinucleotide that starts at a particular position. This can help in modeling CpG or G+C bias.
Positions¶
By default, Segway runs analyses on the whole genome. This can be incredibly time-consuming, especially for training. In reality, training (and even identification) on a smaller proportion of the genome is often sufficient. There are also regions as the genome such as those containing many repetitive sequences, which can cause artifacts in the training process. The --exclude-coords=file and --include-coords=file options specify BED files with regions that should be excluded or included respectively. If both are specified, then inclusions are prcoessed first and the exclusions are then taken out of the included regions.
Warning
BED format uses zero-based half-open coordinates, just like Python. This means that the first nucleotide on chromosome 1 is specified as:
chr1 0 1
The UCSC Genome Browser and Ensembl web interfaces, as well as the wiggle formats use the one-based fully-closed convention, where it is called chr1:1-1.
For example, the ENCODE Data Coordination Center at University of California Santa Cruz keeps the coordinates of the ENCODE pilot regions in this format at <http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/referenceSequences/encodePilotRegions.hg19.bed> (GRCh37/hg19) and <http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/encodeRegions.txt.gz> (NCBI36/hg18). For human whole-genome studies, these regions have nice properties since they mark 1 percent of the genome, and were carefully picked to include a variety of different gene densities, and a number of more limited studies provide data just for these regions. All coordinates are in terms of the GRCh37 assembly of the human reference genome (also called hg19 by UCSC).
After reading in data from a Genomedata archive, and selecting a smaller subset with --exclude-coords and --include-coords, the final included regions are referred to as windows, and are supplied to GMTK for inference. There is no direction connection between the data in different windows during any inference process–the windows are treated independently.
Resolution¶
In order to speed up the inference process, you may downsample the data to a different resolution using the --resolution=res option. This means that Segway will partition the input observations into fixed windows of size res and perform inference on the mean of the observation averaged along the fixed window. This can result in a large speedup at the cost of losing the highest possible precision.
You must use the same resolution in both training and identification.
Model generation¶
Segway generates a model (segway.str) and initial parameters (input.master) appropriate to a dataset using the GMTKL specification language and the GMTK master parameter file format. Both of these are described more fully in the GMTK documentation (cite), and the default structure and starting parameters are described more fully in the Segway article.
The starting parameters are generated using data from the whole genome, which can be quickly found in the Genomedata archive. Even if you are training on a subset of the genome, this information is not used.
You can tell Segway just to generate these files and not to perform any inference using the --dry-run option.
Using --num-instances=starts will generate multiple copies of the input.master file, named input.0.master, input.1.master, and so on, with different randomly picked initial parameters. You may substitute your own input.master files but I recommend starting with a Segway-generated template. This will help avoid some common pitfalls. In particular, if you are going to perform training on your model, you must ensure that the input.master file retains the same #ifdef structure for parameters you wish to train. Otherwise, the values discovered after one round of training will not be used in subsequent rounds, or in the identify or posterior stages.
You can use the --num-labels=labels option to specify the number of segment labels to use in the model (default 2). You can set this to a single number or a range with Python slice notation. For example, --num-labels=5:20:5 will result in 5, 10, and 15 labels being tried. If you specify --num-instances=starts, then there will be starts different instances for each of the labels labels tried.
The question of finding the right number of labels is a difficult one. Mathematical criteria, such as the Bayesian information criterion, would usually suggest using higher numbers of labels. However, the results are difficult for a human to interpret in this case. This is why we usually use ~25 labels for a segmentation of dozens of input tracks. If you use a small number of input tracks you can probably use a smaller number of labels.
There is an experimental --num-sublabels=sublabels option that enables hierarchical segmentation, where each segment label is divided into a number of segment sublabels. This may require some manipulation of model parameter files to actually be useful. The output segmentation will be defined in terms of individual sublabels, where the output label number is equal to the (super) segment label times the number of sublabels per label plus the sublabel number.
Segway allows multiple models of the values of a continuous observation tracks using three different probability distributions: a normal distribution (--distribution=norm), a normal distribution on asinh-transformed data (--distribution=asinh_norm, the default), or a gamma distribution (--distribution=gamma). For gamma distributions, Segway generates initial parameters by converting mean μ and variance σ2 to shape k and scale theta using the equations μ = kθ and σ2 = kθ2. The ideal methodology for setting gamma parameter values is less well-understood, and it also requires an unreleased version of GMTK. I recommend the use of asinh_norm in most cases.
Segment duration model¶
Hard length constraints¶
The --seg-table=file option allows specification of a segment table that specifies minimum and maximum segment lengths for various labels. Here is an example of a segment table:
label len
1:4 200:2200:200
0 200::200
4: 200::200
The header line with label in one column and len in another is mandatory. Slices are specified with a colon as in Python, and are half-open. So label 1:4 specifies labels 1, 2, and 3. For those labels, segment lengths between 200 and 2200 are allowed, with a 200 bp ruler. The ruler for every label must match each other and the option set with --ruler-scale=scale. This may become more free in the future. The ruler is an efficient heuristic that decreases the memory used during inference at the cost of also decreasing the precision with which the segment duration model acts. Essentially, it allows the duration model to switch the behavior of the rest of the model only after a multiple of scale bp has passed. Using 4: for a label means all labels 4 or higher, and they are set to a minimum segment length of 200 and no maximum segment length.
Due to the lack of an epilogue in the model, it is possible to get one segment per sequence that actually does not meet. This is expected and will be fixed in a future release.
Use these segment lengths along with the supervised learning feature with caution. If you try to create something impossible with your supervision labels, such as defining a 2300-bp region to have label 1, which you have already constrained to have a maximum segment length of 2200, GMTK will produce the dreaded zero clique error and your training run will fail. Don’t do this. In practice, due to the imprecision introduced by the 200-bp ruler, a region labeled in the supervision process with label 1 that is only 2000 bp long may also cause the training process to fail with a zero clique error. If this happens either decrease the size of the ruler, increase the size of the maximum segment length, or decrease the size of the supervision region.
Soft length prior¶
There is also a way to add a soft prior on the length distribution, which will tend to make the expected segment length 100000, but will still allow data that strongly suggests another length control. The default expected segment length of 100000 can’t be changed at the moment but will in a future version.
You can control the strength of the prior relative to observed transitions with the --prior-strength=strength option. Setting --prior-strength=1 means there are as many pseudocounts due to the prior as the number of nucleotides in the training regions.
The --segtransition-weight-scale=scale option controls the strength of the prior in another way. It controls the strength of the transition model relative to the data from the observed tracks. The default scale of 1 gives the soft transition model equal strength to a single data track. Using higher or lower values uses comparatively greater or lesser weight to the probability from the soft transition model. Therefore the impact of the prior will be a function of both --segtransition-weight-scale and --prior-strength.
Task selection¶
Segway will perform either (a) model generation and training or (b) identification separately, so it is possible to train on a subset of the genome and identify on the whole thing. To train, use:
segway train GENOMEDATA TRAINDIR
To identify, specify the TRAINDIR you used in the first round:
segway identify GENOMEDATA TRAINDIR IDENTIFYDIR
In both cases, replace GENOMEDATA with the Genomedata archive you’re using. The use of --dry-run will cause Segway to generate appropriate model and observation files but not to actually perform any inference or queue any jobs. This can be useful when troubleshooting a model or task.
Train task¶
Most users will generate the model at training time, but to specify your own model there are the --structure=filename and --input-master=filename options. You can simultaneously run multiple instances of EM training in parallel, specified with the --instances=instances option. Each instance consists of a number of rounds, which are broken down into individual tasks for each training region. The results from each region for a particular instance and round are combined in a quick bundle task. It results in the generation of a parameter file like params.3.params.18 where 3 is the instance index and 18 is the round index. Training for a particular instance continues until at least one of these criteria is met:
- the likelihood from one round is only a small improvement from the previous round; or
- 100 rounds have completed.
Specifically, the “small improvement” is defined in terms of the
current likelihood 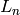 and the log likelihood from the
previous round
and the log likelihood from the
previous round 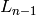 , such that training continues while
, such that training continues while
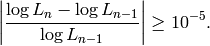
This constant will likely become an option in a future version of Segway.
As EM training produces diminishing returns over time, it is likely that one can obtain acceptably trained parameters well before these criteria are met. Training can be a time-consuming process. You may wish to train only on a subset of your data, as described in Positions.
When all instances are complete, Segway picks the parameter set with the best likelihood and copies it to params.params.
There are two different modes of training available, unsupervised and semisupervised.
Unsupervised training¶
By default, Segway trains in unsupervised mode, which is a form of clustering. In this mode, it tries to find recurring patterns suggested by the data without any additional preconceptions of which regions should be tied together.
Semisupervised training¶
Using the --semisupervised=file option, one can specify a BED file as a list of regions used s supervision labels. The name field of the BED File specifies a label to be enforced during training. For example, with the line:
chr3 400 800 2
one can enforce that those positions will have label 2. You might do this if you had specific reason to believe that these regions were enhancers and wanted to find similar patterns in your data tracks. Using smaller labels first (such as 0) is probably better. Supervision labels are not enforced during the identify task.
To simulate fully supervised training, simply supply supervision labels for the entire training region.
None of the supervision labels can overlap with each other. You should combine any overlapping labels before specifying them to Segway.
General options¶
The --dont-train=file option specifies a file with a newline-delimited list of parameters not to train. By default, this includes the dpmf_always, start_seg, and all GMTK DeterministicCPT parameters. You are unlikely to use this unless you are generating your own models manually.
Recovery¶
Since training can take a long time, this increases the probability that external factors such as a system failure will cause a training run to fail before completion. You can use the --recover=dirname option to specify a previous work directory you’re recovering from.
Identify task¶
The identify mode of Segway uses the Viterbi algorithm to decode the most likely path of segments, given data and a set of parameters, which can come from the train task. Identify runs considerably more quickly than training. While the underlying inference task is very similar, it must be completed on each region of interest only once rather than hundreds of times as in training.
You can either manually set individual input master, parameter, and structure files, or implicitly use the files generated by the train task completed in traindir, and referenced in traindir/train.tab. If you are using training data from an old version of Segway, you must either create a train.tab file or specify the parameters manually, using --structure, --input-master, and --trainable-params.
The --bed=bedfile option specifies where the segmentation should go. If bedfile ends in .gz, then Segway uses gzip compression. The default is segway.bed.gz in the working directory.
You can load the generated BED files into a genome browser. Because the files can be large, I recommend placing them on your web site and supplying the URL to the genome browser rather than uploading the file directly. When using the UCSC genome browser, the bigBed utility may be helpful in speeding access to parts of a segmentation.
The output is in BED format (http://genome.ucsc.edu/FAQ/FAQformat.html#format1), and includes columns for chromosome, start, and end (in zero-based, half-open format), and the label. Other columns to the right are used for display purposes, such as coloring the browser display, and can be safely ignored for further processing. We use colors from ColorBrewer (http://colorbrewer2.org/).
Recovery¶
The --recover=dirname allows recovery from an interrupted identify task. Segway will requeue jobs that never completed before, skipping any windows that have already completed.
Posterior task¶
The posterior inference task of Segway estimates for each position of interest the probability that the model has a particular segment label given the data. This information is delivered in a series of numbered wiggle files, one for each segment label. The individual values will vary from 0 to 100, showing the percentage probability at each position for the label in that file. In most positions, the value will be 0 or 100, and substantially reproduce the Viterbi path determined from the identify task.
Posterior results can be useful in determining regions of ambiguous labeling or in diagnosing new models. The mostly binary nature of the posterior assignments is a consequence of the design of the default Segway model, and it is possible to design a model that does not have this feature. Doing so is left as an exercise to the reader.
You may find you need to convert the bedGraph files to bigWig format first to allow small portions to be uploaded to a genome browser piecewise.
Creating layered output¶
Segway produces BED Files as output with the segment label in the name field. While this is the most sensible way of interchanging the segmentation with other programs, it can be difficult to visualize. We supply a segway-layer program that transforms segmentation BED files into “layered” BED files with rows for each possible Segment label and thick boxes at the location of each label. This is what we show in the screenshot figure of the Segway article. This is much easier to see at low levels of magnification. The layers are also labeled, removing the need to distinguish them exclusively by color. segway-layer supports the use of standard input and output by using - as a filename, following a common Unix convention. Segway automatically runs segway-layer at the end of an identify task.
The mnemonic files used by Segway and Segtools have a simple format. They are tab-delimited files with a header that has the following columns: old, new, and description. The old column specifies the original label in the BED file, which is always produced as an integer by Segway. The new column allows the specification of a short alphanumeric mnemonic for the label. The description column is unused by segway-layer, but you can use it to add helpful annotations for humans examining the list of labels, or to save label mnemonics you used previously. The row order of the mnemonic file matters, as the layers will be laid down in a similar order. Mnemonics sharing the same alphabetical prefix (for example, A0 and A1) or characters before a period (for example, 0.0 and 0.1) will be rendered with the same color.
segtools-gmtk-parameters in the Segtools package can automatically identify an initial hierarchical labeling of segmentation parameters. This can be very useful as a first approximation of assigning meaning to segment labels.
A simple mnemonic file appears below:
old new description
0 TSS transcription start site
2 GE gene end
1 D dead zone
Technical matters¶
Working files¶
Segway must create a number of working files in order to accomplish its tasks, and it does this in the directory specified by the required workdir argument.
The observation files can be quite large, taking up 8 bytes per track per position and cannot be compressed. Since they are needed multiple times during training, they are generated in the working directory. As an exception, if you turn off training and specify only the identify task, the files will be generated independently on temproary space on each machine. This is because otherwise they could take terabytes for identifying on the whole human genome with dozens of tracks.
To change the location of the observation files, use the --observations=dir option. This can be helpful when you plan to conduct multiple training tasks on the same data but with different parameters. Be careful to finish observation writing for at least one task first, so all the observations will be written out, before trying to start the next task. If you fail to do this, race conditions may lead to undefined effects. Also, all data selection options (--track, --include-coords, --exclude-coords) must be exactly the same when sharing observation files. Otherwise you are likely to get unexplained failures.
You will find a full description of all the working files in the Files section
Distributed computing¶
Segway can currently perform the training, identification, and posterior tasks only using a cluster controllable with the DRMAA interface. I have only tested it against Sun Grid Engine and Platform LSF, but it should be possible to work with other DRMAA-compatible distributed computing systems, such as PBS Pro, PBS/TORQUE, Condor, or GridWay. If you are interested in using one of these systems, please contact Michael so he correct all the fine details. A standalone version is planned, but for now you must have a clustering system. Try installing the free SGE on your workstation if you want to run Segway without a full clustering system.
The --cluster-opt option allows the specification of native options to your clustering system—those options you might pass to qsub (SGE) or bsub (LSF).
Memory usage¶
Inference on complex models or long sequences can be memory-intensive. In order to work efficiently when it is not always easy to predict memory use in advance, Segway controls the memory use of its subtasks on a cluster with a trial-and-error approach. It will submit jobs to your clustering system specifying the amount of memory they are expected to take up. Your clustering system should allocate these jobs such that the amount of memory on one host is not overcommitted. If a job takes up more memory than allocated, then it will be killed and restarted with a larger amount of memory allocated, along the progression specified in gibibytes by --mem-usage=progression. The default progression is 2,3,4,6,8,10,12,14,15.
We usually train on regions of no more than 2,000,000 frames, where a single frame contains the number of nucleotides set by the --resolution option. If you use more than that many, GMTK might run out of dynamic range. This manifests itself as a “zero clique error.” Identify mode rescales probabilities at every frame so that this is not a problem. However, you will probably want to split the input sequences somewhat because larger sequences make more difficult work units (greater memory and run time costs) and thereby impede efficient parallelization. The --split-sequences=size option will split up sequences into windows with size frames each. The default size is 2,000,000. Decreasing to 500,000 will greatly improve speed at the cost of more artefacts at split boundaries.
Reporting¶
Segway produces a number of logs of its activity during tasks, which can be useful for analyzing its performance or troubleshooting. These are all in the workdir/log directory.
Shell scripts¶
Segway produces three shell scripts in the log directory that you can use to replay its subtasks at different levels of abstractions. The top-level segway.sh records the command line used to run Segway. The run.sh script gives you the GMTK commands called by Segway. A small number of these are still produced when --dry-run is specified. The details.sh script contains the exact commands dispatched by Segway, including wrapper commands that monitor memory usage, create and delete local temporary files with observation data, and convert GMTK’s output ot BED, among other things.
Summary reports¶
The jobs.tab file contains a tab-delimited file with each job Segway dispatched in a different row, reporting on job identifier (jobid), job name (jobname), GMTK program (prog), number of segment labels (num_segs), number of frames (num_frames), maximum memory usage (maxvmem), CPU time (cpu) and exit/error status (exit_status). Jobs are written as they are completed. The exit status is useful for determining whether the job succeeded (status 0) or failed (any other value, which is sometimes numeric, and sometimes text, depending on the clustering system used).
The likelihood.*.tab files each track the progression of likelihood during a single instance of EM training. The file has a single column, one for each round of training, which contains the log likelihood. More positive values are better.
GMTK reports¶
The jt_info.txt and jt_info.posterior.txt files describe how GMTK builds a junction tree. It is of interest primarily during GMTK troubleshooting. You are unlikely to use it.
Task output¶
The output directory contains the output of the actual GMTK commands run by Segway. The o directory contains standard output and the e directory contains standard error. If a job fails and repeats, the output from the new job is appended to the old. The --verbosity=verbosity option controls how much diagnostic information that GMTK writes into these files. The default and minimum value is 0. Raise this value for more information, and see the GMTK documentation for a description of various levels of verbosity. Setting verbosity=30 can be particularly helpful in diagnosing model problems. Keep in mind that very high values (above 60) will produce tons of output—maybe terabytes.
Performance¶
Some factors that affect compute time and memory requirements:
- the length of the longest region you are training or identifying on
- the number of tracks
- the number of labels
The longest region forms a bottleneck during training because Segway cannot start the next round of training before all regions in the previous round are done. So if you specify three regions, one of which is 10 Mbp long, and the other are 100 kbp, the 10 Mbp region is going to be a limiting factor. You can use --split-sequences (see above) to put an upper bound on region size.
Troubleshooting¶
When Segway reports “end of memory progression reached without success”, this usually means that dispatched GMTK tasks failed repeatedly. Look in the output/e files to see what the cause of the underlying error was. Also log/jobs.tab lists all the jobs and whether they reported an error in the form of nonzero exit status.
Are your bundle jobs failing? This might be because an accumulator file (written by individual job) is corrupted or truncated. This can happen when you run out of disk space.
If it is not immediately apparent why a job is failing, it is probably useful to look in log/run.sh to find the command line that Segway uses to call GMTK. Try running that to see if it gives you any clues. You may want to switch the GMTK option -verbosity 0 to -verbosity 30 to get more information.
An error like:
ERROR: discrete observed random variable 'presence_dnase', frame 0, line 23, specifies a feature element 14:14 that is out of discrete range ([23:45] inclusive) of observation matrix
probably indicates that you are incorrectly mixing and matching train.tab files and segway.str files from different training runs.
/net/noble/vol2/home/avinash/shortcuts/multipass/segway_src/runs/2011/0413-segway-coordinated-jan2011/k562.coordinated/t20110414/
Names used by Segway¶
Workdir files¶
Segway expects to be able to create many of these files anew. To avoid data loss, by default, it will quit if they already exist. If you use the --clobber option, Segway will overwrite the whole workdir instead.
| Filename | Description |
|---|---|
| accumulators/ | intermediate files used to pass E-step results to the M-step of EM training |
| → acc.*.bin | accumulator for a particular instance and region (reused each round) |
| auxiliary/ | miscelaneous model files |
| → dont_train.list | defines list of hidden random variables that are not trained |
| → segway.inc | C preprocessor (cpp) include file used in structure |
| likelihood/ | GMTK’s report of the log likelihood for the most recent M-step of EM training |
| → likelihood.*.ll | contains text of the last log likelihood value for an instance. Segway uses this to decide when to stop training |
| log/ | diagnostic information |
| → details.sh | script file that includes the exact command-lines queued by Segway, with wrapper scripts |
| → jobs.tab | tab-delimeted summary of jobs queued, including resource informatoin and exit status |
| → jt_info.txt | log file used by GMTK when creating a junction tree |
| → jt_info.posterior.txt | log file used by GMTK when creating a junction tree in posterior mode |
| → likelihood.*.tab | tab-delimited summary of likelihood by training instance; can be used to examine how fast training converges |
| → run.sh | list of commands run by Segway, not including wrappers that create and clean up temporary files such as observations used during identification |
| → segway.sh | reports the command-line used to run Segway itself |
| observations/ | decompressed, and potentially large raw observation files created from a Genomedata archive located elsewhere |
| → *.*.float32 | continuous data for a particular region |
| → *.*.int | indicator data (present/absent) for a particular region |
| → float32.list | list of continuous data files |
| → int.list | list of indicator data files |
| → observations.tab | tab-delimited description of observations used|rarr| Used to check that an existing directory specified with --observations matches the data specified at the command-line |
| output/ | diagnostic output of individual GMTK jobs |
| output/e/ | stderr |
| output/e/0,1,... | stderr for a particular training instance (0, 1, ...) |
| output/e/identify | stderr for identification |
| output/o/ | stdout |
| params/ | generated and trained parameters for a given instance |
| → input.*.master | generated hyperparameters and starting parameters |
| → input.master | best set of hyperparameters and starting parameters |
| → params.*.params.* | trained parameters for a given instance and round |
| → params.*.params | final trained parameters for a given instance |
| → params.params | best final set of trained parameters |
| segway.bed.gz | segmentation in BED format |
| segway.str | dynamic Bayesian network structure |
| train.tab | important file locations and hyperparameters used in training, to be passed to identify |
| triangulation/ | triangulation files used for DBN interface |
| → segway.str.*.*.trifile | triangulation file |
| viterbi/ | intermediate BED files created during distributed Viterbi decoding, which get merged into segway.bed.gz |
Job names¶
In order to watch Segway’s progress on your cluster, it is helpful to understand how it names jobs. A job name for the training task might look like this:
emt0.1.34.traindir.ed03201cea2047399d4cbcc4b62f9827
In this example, emt means expectation maximization training, the 0 means instance 0, the 1 means round 1, and the 34 means window 34. The name of the training directory is traindir, and ed03201cea2047399d4cbcc4b62f9827 is a universally unique identifier for this particular Segway run. This can be useful if you want to manage all of your jobs on your clustering system with wildcard specification. On SGE you can delete all the jobs from this run with:
qdel "*.ed03201cea2047399d4cbcc4b62f9827"
On LSF, use:
bkill -J "*.ed03201cea2047399d4cbcc4b62f9827"
Jobs created in the identify (vit) task are named similarly:
vit34.identifydir.4f32630d53724f08b34a8fc58793307d
Of course, there are no instances or rounds for the identify task, so only the sequence index is reported.
Tracks¶
Tracks are named according to their name in the Genomedata archive. For GMTK internal use, periods are converted to underscores. There is a special track name dinucleotide. Use this track name to model a dinucleotide symbol at each position with a 16-symbol multionmial distribution specified in the form of a conditional probability table.
Python interface¶
I have designed Segway such that eventually one may call different components directly from within Python.
You can then call the appropriate module through its main() function with the same arguments you would use at the command line. For example:
from segway import run
GENOMEDATA_DIRNAME = "genomedata"
run.main(["--random-starts=3", "train", GENOMEDATA_DIRNAME])
All other interfaces (the ones that do not use a main() function) to Segway code are undocumented and should not be used. If you do use them, know that the API may change at any time without notice.
Support¶
For support of Segway, please write to the <segway-users@uw.edu> mailing list, rather than writing the authors directly. Using the mailing list will get your question answered more quickly. It also allows us to pool knowledge and reduce getting the same inquiries over and over. You can subscribe here:
https://mailman1.u.washington.edu/mailman/listinfo/segway-users
Specifically, if you want to report a bug or request a feature, please do so using the Segway issue tracker at:
http://code.google.com/p/segway-genome/issues/
If you do not want to read discussions about other people’s use of Segway, but would like to hear about new releases and other important information, please subscribe to <segway-announce@uw.edu> by visiting this web page:
https://mailman1.u.washington.edu/mailman/listinfo/segway-announce
Announcements of this nature are sent to both segway-users and segway-announce.
Command-line usage summary¶
All programs in the Segway distribution will report a brief synopsis of expected arguments and options when the --help option is specified and version information when --version is specified.
Usage: segway [OPTION]... TASK GENOMEDATA TRAINDIR [IDENTIFYDIR]
- Options:
--version show program’s version number and exit -h, --help show this help message and exit - Data selection:
-t TRACK, --track=TRACK append TRACK to list of tracks to use (default all) --tracks-from=FILE append tracks from newline-delimited FILE to list of tracks to use --include-coords=FILE limit to genomic coordinates in FILE (default all) --exclude-coords=FILE filter out genomic coordinates in FILE (default none) --resolution=RES downsample to every RES bp (default 1) - Model files:
-i FILE, --input-master=FILE use or create input master in FILE (default WORKDIR/params/input.master) -s FILE, --structure=FILE use or create structure in FILE (default WORKDIR/segway.str) -p FILE, --trainable-params=FILE use or create trainable parameters in FILE (default WORKDIR/params/params.params) --dont-train=FILE use FILE as list of parameters not to train (default WORKDIR/auxiliary/dont_train.list) --seg-table=FILE load segment hyperparameters from FILE (default none) --semisupervised=FILE semisupervised segmentation with labels in FILE (default none) - Intermediate files:
-o DIR, --observations=DIR use or create observations in DIR (default WORKDIR/observations) -r DIR, --recover=DIR continue from interrupted run in DIR - Output files:
-b FILE, --bed=FILE create identification BED track in FILE (default WORKDIR/segway.bed.gz) - Modeling variables:
-D DIST, --distribution=DIST use DIST distribution (default asinh_norm) --num-instances=NUM run NUM training instances, randomizing start parameters NUM times (default 1) -N SLICE, --num-labels=SLICE make SLICE segment labels (default 2) --num-sublabels=NUM make NUM segment sublabels (default 1) --max-train-rounds=NUM each training instance runs a maximum of NUM rounds (default 100) --ruler-scale=SCALE ruler marking every SCALE bp (default 10) --prior-strength=RATIO use RATIO times the number of data counts as the number of pseudocounts for the segment length prior (default 0.000000) --segtransition-weight-scale=SCALE exponent for segment transition probability (default 1.000000) - Technical variables:
-m PROGRESSION, --mem-usage=PROGRESSION try each float in PROGRESSION as the number of gibibytes of memory to allocate in turn (default 2,3,4,6,8,10,12,14,15) -S SIZE, --split-sequences=SIZE split up sequences that are larger than SIZE bp (default 2000000) -v NUM, --verbosity=NUM show messages with verbosity NUM (default 0) --cluster-opt=OPT specify an option to be passed to the cluster manager - Flags:
-c, --clobber delete any preexisting files -n, --dry-run write all files, but do not run any executables
Utilities¶
Usage: segway-layer [OPTION]... [INFILE] [OUTFILE]
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -m FILE, --mnemonic-file=FILE specify tab-delimited file with mnemonic replacement identifiers for segment labels -s <ATTR VALUE>, --track-line-set=<ATTR VALUE> set ATTR to VALUE in track line
Usage: segway-winner [OPTION]... DIR
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i, --input-master print input master file name -p, --params print parameters file name -c, --copy copy files to final winning file locations --clobber overwrite existing files
Helpful commands¶
Here are some short bash scripts or one-liners that are useful:
Make a tarball of parameters and models from various directories:
(for DIR in traindir1 traindir2; do
echo $DIR/{auxiliary,params/input.master,params/params.params,segway.str,triangulation}
done) | xargs tar zcvf training.params.tar.gz
Rsync parameters from $REMOTEDIR on $REMOTEHOST to $LOCALDIR:
rsync -rtvz --exclude output --exclude posterior --exclude viterbi --exclude observations --exclude "*.observations" --exclude accumulators $REMOTEHOST:$REMOTEDIR $LOCALDIR
Print all last likelihoods:
for X in likelihood.*.tab; do dc -e "8 k $(tail -n 2 $X | cut -f 1 | xargs echo | sed -e 's/-//g') sc sl ll lc - ll / p"; done
Recover as much as possible from an incomplete identification run without completing it. Note that this does not combine adjacent lines of same segment. BEDTools might be able to do this for you. You will have to create your own header.txt with appropriate track lines:
cat header.txt <(find viterbi -type f | sort | xargs cat) | gzip -c > segway.bed.gz