https://github.com/Noble-Lab/pastis-protocol
The purpose of this web page is to describe the data files that accompany the project that are hosted online, in addition to explain how to modify some of the scripts.
Chromosome conformation capture methods such as Hi-C provide rich information about the three-dimensional configuration of DNA in a population of cells. This data is most frequently visualized using heatmap representations of the 2D locus-to-locus contact map. In practice, however, projecting the DNA into a three-dimensional representation can offer valuable intuitions and insights that are not always easy to glean from the contact map. The PASTIS software infers, for a given Hi-C contact map, a corresponding consensus 3D structure, where each bead in the structure corresponds to one row (or column) of the Hi-C map. The algorithm models the distances between beads in the struture using a poisson likelihood function and is able to generate full-genome structures of haploid or diploid structures. PASTIS is implemented in Python and requires only basic knowledge of the command line interface on Linux, Windows, or MacOS. In this protocol, we demonstrate how to use PASTIS to infer 3D structures from Hi-C matrices derived from yeast and human samples. We also show how to visualize the resulting structures in various ways, including direct viewing with Python tools, via several PDB viewers, and using two different genome browsers (4D Nucleome Browser and WashU Epigenome Browser).
| File | Filesize | Description |
|---|---|---|
| 4DNFII84FBKM.matrix | 141 MB | Counts matrix used in the protocol. Extracted from 4DNFII84FBKM.hic from 4DN database |
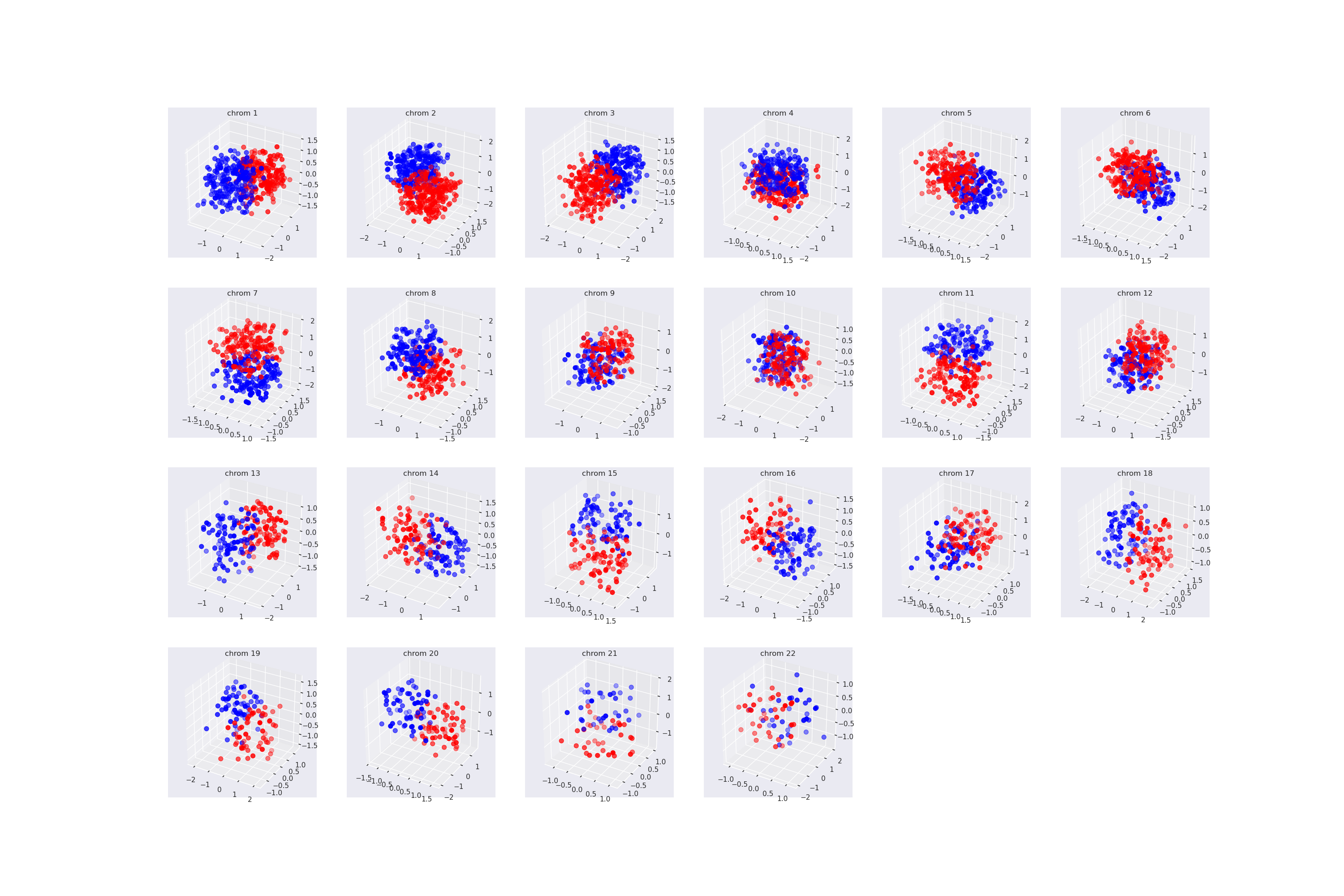
| chroms_structure_mpl.png | 1320 KB | Plot of chromosomes generated in protocol. |
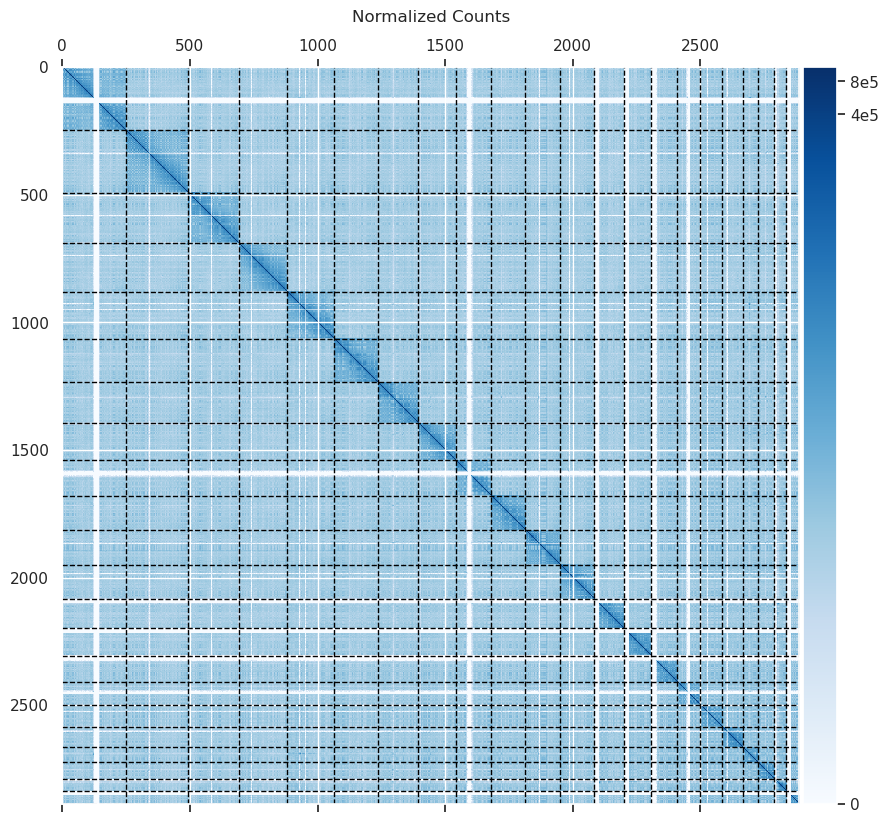
| counts_heatmap.png | 997 KB | Counts heatmap generated during protocol. |
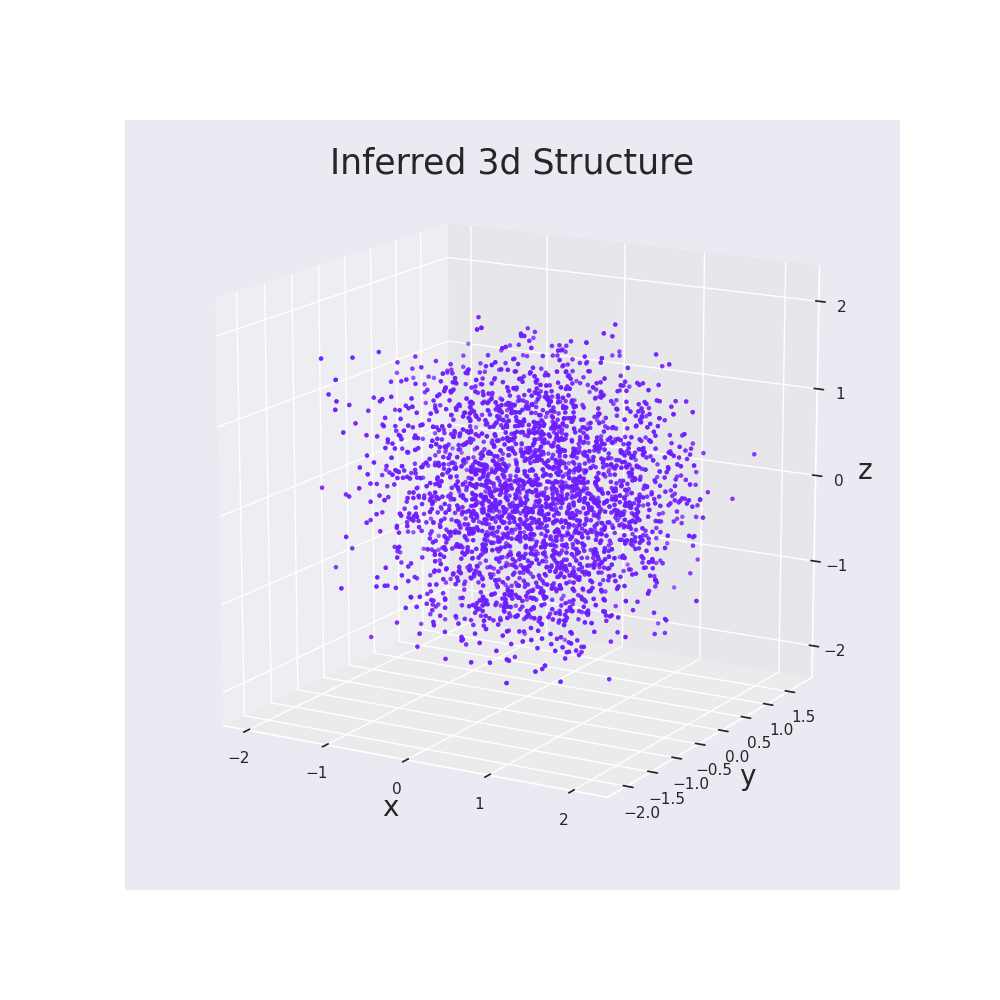
| full_structure_mpl.png | 215 KB | Full structure plot generated during protocol. |
| hg38AB.chrom.sizes | 716 B | Chromosome sizes file generated from data/hg38.chrom.sizes during protocol with homologs labeled as A / B. |
| hg38_notAB.chrom.sizes | 336 B | Chromosome sizes file generated from data/hg38.chrom.sizes during protocol without homologs labeled separately. |
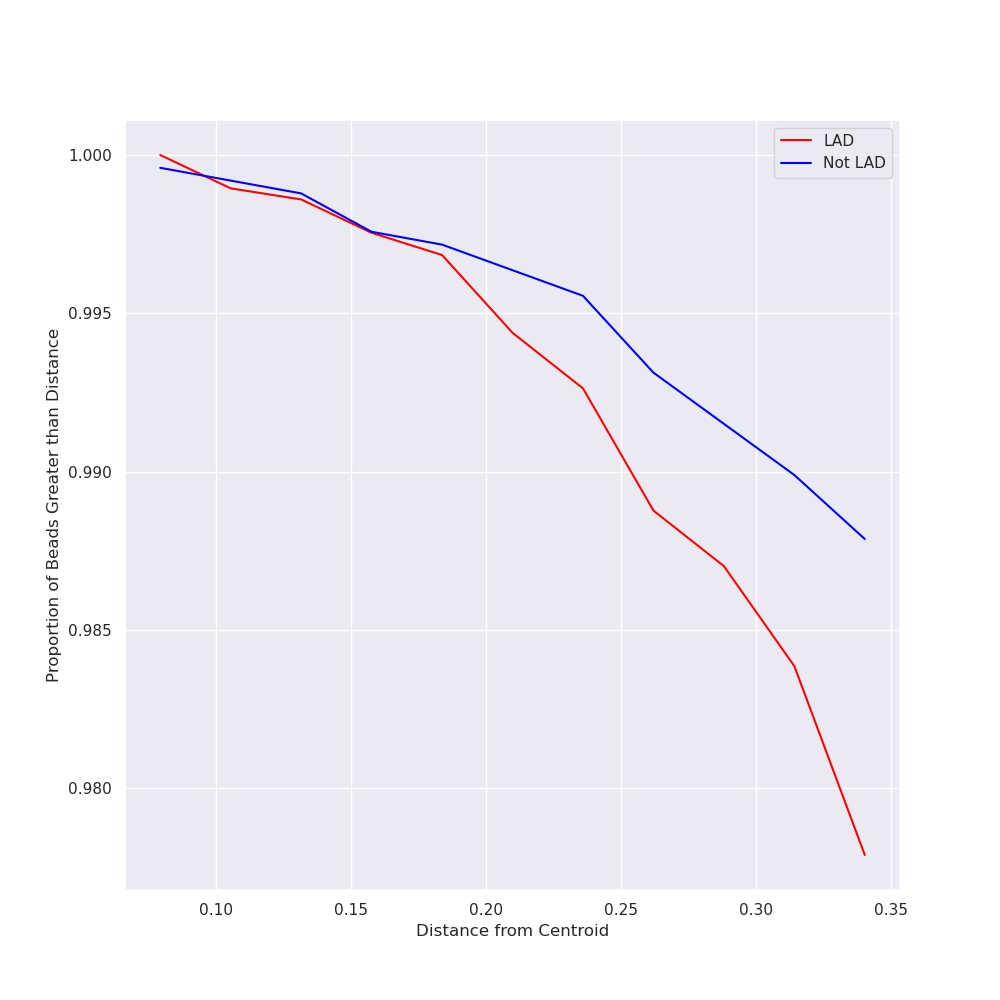
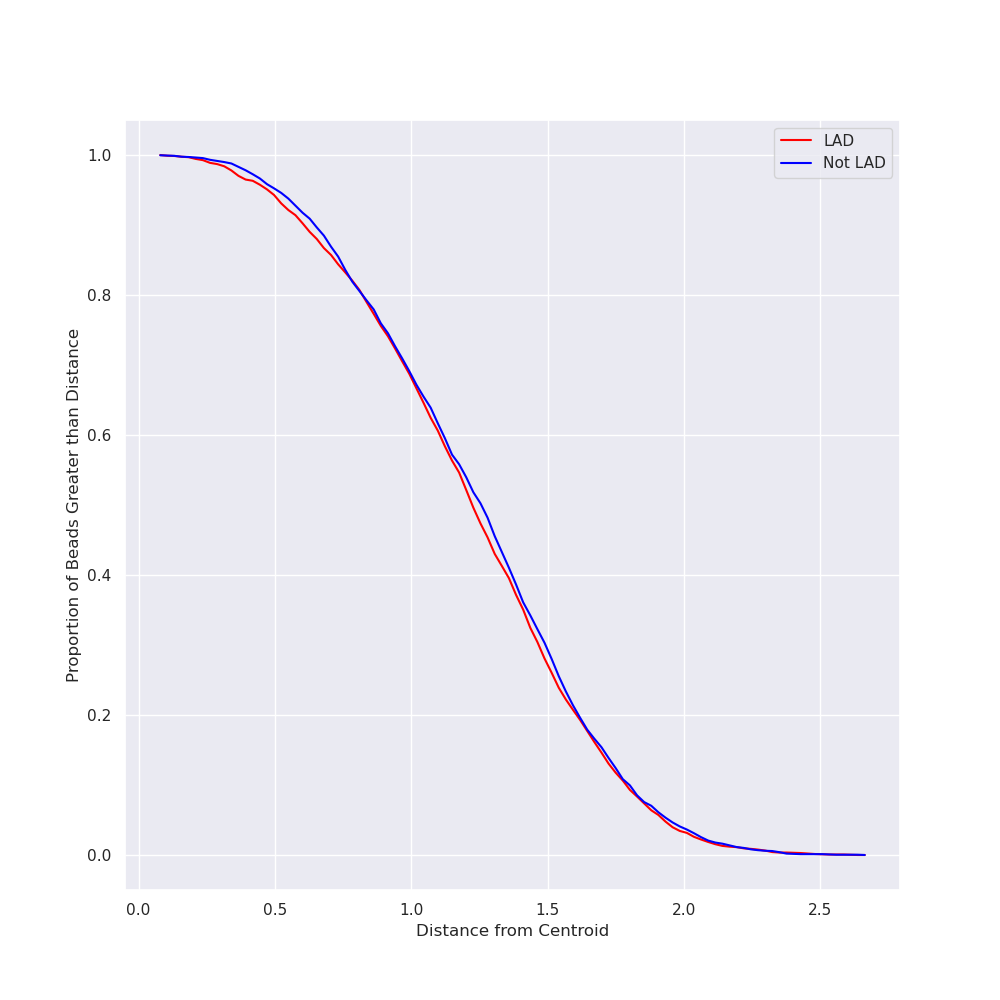
| lad_distances_cut.png | 57 KB | Zoomed in LAD distances plot generated during protocol. |
| lad_distances_full.png | 57 KB | Zoomed in LAD distances plot generated during protocol. |
| lad_notlad_AB.bedGraph | 165 KB | bedGraph file denoting LAD and not LAD regions of structure with homologs labeled as A / B. |
| lad_notlad_AB.bw | 60 KB | bigWig file generated from lad_notlad_AB.bedGraph and hg38AB.chrom.sizes denoting LAD and not LAD regions of structure with homologs labeled as A / B. |
| lad_notlad.bedGraph | 80 KB | bedGraph file denoting LAD and not LAD regions of structure with homologs not labeled separately. |
| lad_notlad.bw | 43 KB | bigWig file generated from lad_notlad.bedGraph and hg38_notAB.chrom.sizes denoting LAD and not LAD regions of structure with homologs not labeled separately. |
| struct_inferred.000.coords | 403 KB | Structure generated by PASTIS during protocol. |
| struct_inferred.000.g3d | 193 KB | struct_inferred.000.coords converted to g3d format. |
| struct_inferred.000.nucle3d | 332 KB | struct_inferred.000.coords converted to nucle3d format. |
| struct_inferred.000.pdb | 369 KB | struct_inferred.000.coords converted to PDB format. |
To use a different counts matrix, I will suppose you have chosen a matrix from the 4DN database.
You will not have to use a file other than data/hg38.chrom.sizes unless you are not working with the human genome anymore. If this is the case, you will have to do a few things:
After you do the aforementioned modifications and run PASTIS, I recommend making sure the call to "run_pastis.sh" in bin/run_all.sh is COMMENTED OUT (unless you want to re-run PASTIS). Then, run bin/run_all.sh to regenerate result plots and result files (ie PDB files, bigWig files, etc.). This is the easiest way to regenerate results. Furthermore, to generate the genome browser figures, you will have to re-screenshot them and upload them into the manuscript.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}