This is a guide to downloading and using Fido, a method for protein identification in MS/MS proteomics. Think of it like a protein delivery dog: you bring it the scored matches between peptides and spectra, and it fetches a list of proteins ranked by posterior probability by doing clever tricks.
Note: The version of Fido distributed here is no longer maintained. Fido has now been integrated into Percolator, which is distributed as stand-alone software and as part of the Crux mass spectrometry analysis toolkit.

Running Fido (from the command line)
To run Fido, type
usage: bin/FidoChooseParameters [-p] [-a] [-g] [-c <n>] <graph_file> <targetDecoy_file> bin/FidoChooseParameters [-p] [-a] [-g] [-c <n>] <graph_file> <targetDecoy_file> <l1> bin/FidoChooseParameters [-p] [-a] [-g] [-c <n>] <graph_file> <targetDecoy_file> <l1> <l2> where l1 is the log2 of maximum number of subgraph connected states, l2 is the log2 for the main calculation, and l1 is only used for precalculation option -p omits cleaning the peptide names, option -a uses all PSM matches instead the best one, and option -g uses protein group level inference. option -c sets start parameter's accurary level (1-3) 1 = best / slower 2 = relaxed / faster 3 = sloppy / very fastwhere psm_graph_file is the path to your peptide spectrum match (PSM) graph file and targetDecoy.txt is a file with two lines, the first all targets (i.e. true accessions) and the second all decoys (i.e. false positive accessions). An example is shown here.
If you want to choose your parameters yourself with some other criteria, use:
bin/Fido psm_graph_file prior_probability alpha_probability beta_probabilityor
bin/Fido psm_graph_file prior_probability alpha_probability beta_probability log2_maximum_number_of_states
where psm_graph_file is the path to your peptide spectrum match (PSM) graph file, and prior_probability, alpha_probability, and beta_probability are values for gamma (the protein prior probability), alpha (the peptide emission probability), and beta (the spurious peptide identification probability), respectively. Reasonable choices for these parameters (both by accuracy and calibration) are gamma = 0.5, alpha = 0.1, and beta = 0.01, but you should choose the best parameters for your data set using the FidoChooseParameters command.
The optional parameter log2_maximum_number_of_states will be used to split up problems that are intractable to solve exactly; the smaller it is set, the faster it will be, but at the cost of accuracy. If 4 is used, then it will ensure that every connected subgraph only takes 16 or fewer steps to marginalize; if 10 is used, then it will ensure every connected subgraph takes 1024 or fewer steps to marginalize.
Input File Format
The format for the PSM graph file is this:e peptide_string
r protein that would create this peptide using theoretical digest
r second protein
...
r final protein
p probability of the peptide match to the spectrum (given by PeptideProphet or comparable).
e peptide_string
r protein that would create this peptide using theoretical digest
r second protein
...
r final protein
p probability of the peptide match to the spectrum (given by PeptideProphet or comparable).
...
For example, this graph
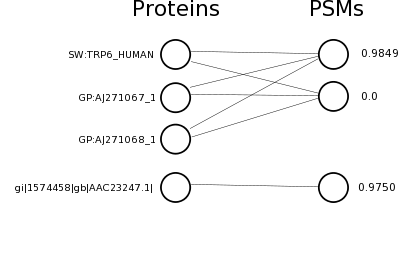
would correspond to this PSM graph file:
e EEEMPEPK r SW:TRP6_HUMAN r GP:AJ271067_1 r GP:AJ271068_1 p 0.9849 e LLEIIQVR r SW:TRP6_HUMAN r GP:AJ271067_1 r GP:AJ271068_1 p 0.0 e FAFNNKPNLEWNWK r gi|1574458|gb|AAC23247.1| p 0.9750
Output File Format
The ouput of Fido is a column of posteriors (sorted in descending order) and the proteins that they correspond to:
0.9988 { gi|1574458|gb|AAC23247 }
0.6788 { SW:TRP6_HUMAN , GP:AJ271067_1 , GP:AJ271068_1 }
where all proteins on the line get the same score. The immediately
above output is equivalent to this:
0.9988 { gi|1574458|gb|AAC23247 }
0.6788 { SW:TRP6_HUMAN }
0.6788 { GP:AJ271067_1 }
0.6788 { GP:AJ271068_1 }
It should be noted that not all proteins receiving the same score will necessarily be put on the same line. This is extremely important for computing ROC curves correctly when some proteins receive the same posterior.
Reformatting PepXML (PeptideProphet output)
To produce the input file format from a pepXML file, interact.xml run:
xsltproc src/xsl/pepProph2Pivdo.xsl input.xml
The output will be the properly formatted PSM graph file. Problems using this xsl code are (thus far) namespace related.
Downloading and Building Fido
Fido has been compiled under g++ 4.x, but with older versions of g++, it may need a minimal amount of tweaking.
Download the C++ version of Fido here.
Download the Python versio of Fido here.
How to Cite Fido
Please see the J. of Proteome Research link here
A License to ID Proteins
Fido is offered under an MIT license (free). A detailed license, rich in legalese, is included in the source code download. Short explanation: use Fido, mention me, get rich, no problem. Woo!